
Research topics
Current projects
The Arab genome project
Discoveries from the human genome, HapMap, and 1000 genome projects have collectively contributed toward the creation of a catalog of human genetic variations that has improved our understanding of human diversity. Despite the collegial nature of many of these genome study consortiums, which has led to the cataloging of genetic variations of different ethnic groups from around the world, genome data on the Arab population remains overwhelmingly underrepresented.
The National Arab Genome project in the United Arab Emirates (UAE) aims to address this deficiency by using Next Generation Sequencing (NGS) technology to provide data to improve our understanding of the Arab genome and catalog variants that are unique to the Arab population of the UAE. The project was conceived to shed light on the similarities and differences between the Arab genome and those of the other ethnic groups.

The Animals Genome Assembly project
Obtaining the reference genome sequence for several organisms (including Camal, Horse and Falcon) by De novo Assembly often using illumina technology.
Genome assembly and genome annotation are areas where there are no gold standards. Projects are often explorative, and knowing if your results are good or bad is often hard to determine. This is especially true if you are working with organisms only distantly related to already sequenced ones, which leaves you with little to compare with.
Try to set an aim with your study, and stop working with the assembly and annotation once you have a result that allows you to reach that aim. Do not fall into the trap of wanting a “perfect” genome, as this tends to lead to a project that never ends. But also do not be afraid to start your own assembly and annotation project.
With the development of new sequencing technologies it is more feasible than ever, and a well assembled and annotated genome will be a resource you can use for many years to follow.
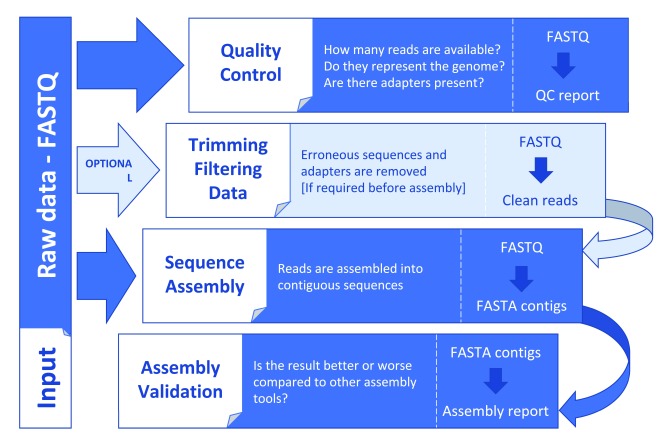
The Transcriptome Explore-EXPRESS Viewer project
With the advances of RNA sequencing technologies, scientists need new tools to analyze transcriptome data. We introduce TEE-Viewer, a new visualization tool dedicated to transcriptomic data. The program offers simple ways to represent transcriptome data for single or multiple samples.
It is a handy tool for scientists who use RNA-Seq/Microarray data to compare multiple transcriptomes, for example, to compare gene expression and alternative splicing of cancer samples or of different development stages.
Past projects
The BLUEPRINT project
The BLUEPRINT Consortium has been formed with the aim of generating at least 100 reference epigenomes and studying them to advance and exploit knowledge of the underlying biological processes and mechanisms in health and disease. BLUEPRINT will focus on distinct types of haematopoietic cells from healthy individuals and on their malignant leukaemic counterparts. Reference epigenomes will be generated by state-of-the-art technologies from highly purified cells for a comprehensive set of epigenetic marks in accordance with quality standards set by the International Human Epigeneome Consortium (IHEC). This resource-generating activity will be conducted at dedicated centres to be complemented by confederated hypothesis-driven research into blood-based diseases, including common leukaemias and autoimmune disease (T1D), by epigenetic targets and compound identification, and by discovery and validation of epigenetic markers for diagnostic use. Key to the success of BLUEPRINT will be the integration with other data sources (i.e. ICGC, 1000 genomes and ENCODE), comprehensive bioinformatic analysis, and user-friendly dissemination to the wider scientific community. The involvement of innovative companies will energize epigenomic research in the private sector by creating new targets for compounds and the development of smart technologies for better diagnostic tests.
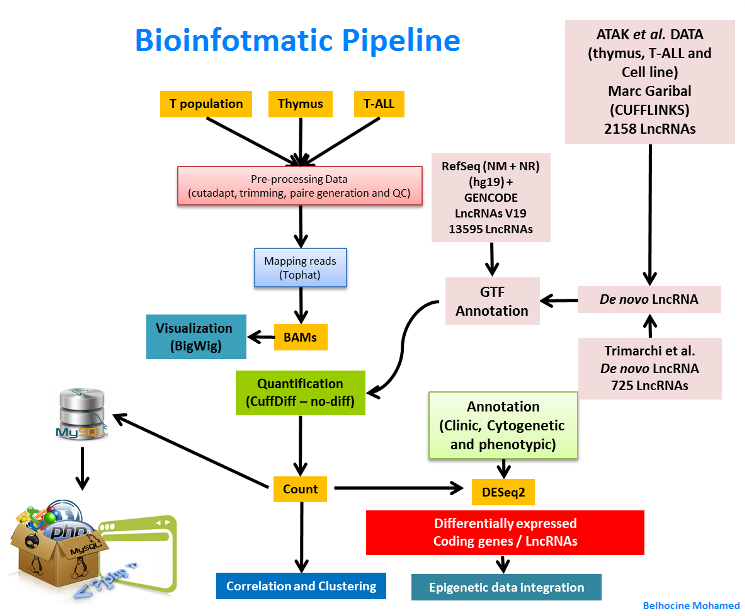
LincRNA project
Transcription of essentially the entire eukayotic genome generates a myriad of non-coding RNA species that show complex overlapping patterns of expression and regulation. Of those, the so-called Long Intergenic NonCoding RNAs (lincRNAs) are among the least well-understood transcript species. Although, only few LincRNAs has been fully characterized to date, they are suggested to play important roles in transcriptional and epigenetic regulation of gene expression, and diseases. Here, we propose to characterize LincRNAs important for T-cell differentiation and potentially relevant in T-cell acute lymphoblastic leukemia (T-ALL). First, we will identify and define the complete set of LincRNAs expressed at key stages of mouse and human T cell differentiation. This will be achieved by performing RNA-Seq and ChIP-Seq experiments. Secondly, a set of differentially expressed LincRNAs will be selected for thorough functional characterization, including (1) Analyses of LincRNAs expression in other tissues; (2) knock-down and/or overexpression in primary thymocytes; and (3) Analyses of their expression in large collections of T-ALLs and correlation with their clinical outcome.